




De mate van aantrekking in waterstofbruggen tussen twee moleculen wordt ook beïnvloed door de overige atomen in dat molecuul, blijkt uit Spaans-Nederlands onderzoek in Physical Chemistry Chemical Physics.
De waterstofbrug of waterstofbinding speelt een grote rol in de interactie tussen biomoleculen als DNA en eiwitten, waar het zelfassemblage in de hand speelt. Wil je dat principe zelf kunnen benutten, dan is het zaak dat je waterstofbruggen goed begrijpt. Daar helpt recent onderzoek bij van Celine Nieuwland, Célia Fonseca Guerra en collega’s van de VU Amsterdam en de Universiteit van Barcelona, dat kijkt naar de invloed van de moleculaire backbone van waterstofbindende moleculen.
Normaal gesproken kijk je alleen naar de interacties die zich ‘op de voorgrond’ afspelen, dus tussen de deels elektropositieve waterstofatomen (H-O en H-N) en elektronegatieve atomen als zuurstof en stikstof. Dat het gangbare waterstofbrugmodel niet juist is, hebben we al eens eerder besproken. Daarop voortbordurend wilden de onderzoekers met dit werk het concept dat je waterstofbrugsterkte kunt verklaren met enkel de zogenoemde frontier (H—N/O)-atomen verder aan de tand voelen.
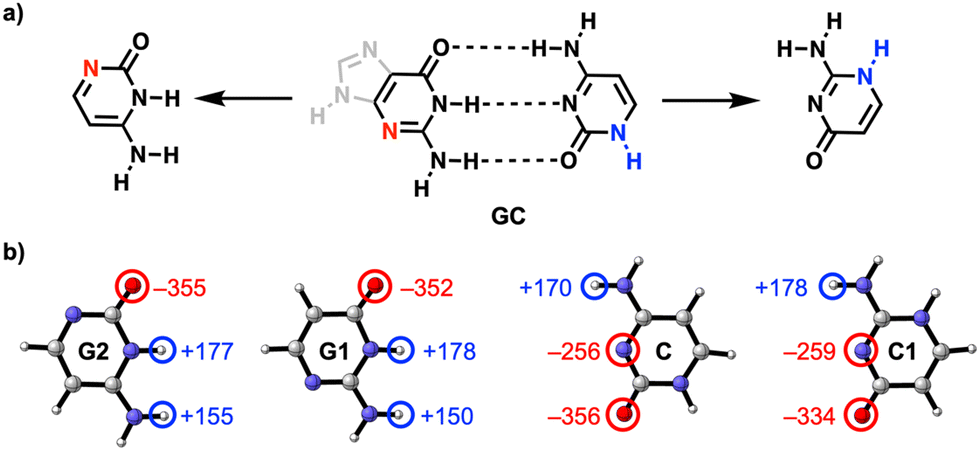
Dat deden ze door computationeel te kijken naar de waterstofbindingen tussen de relevante delen van guanine (G) en cytosine (C), twee moleculen die je ook in DNA tegenkomt waar ze een complementair basepaar vormen. Beide moleculen hebben een stikstofatoom in de ring zitten dat niet direct meedoet in de waterstofbindingen.
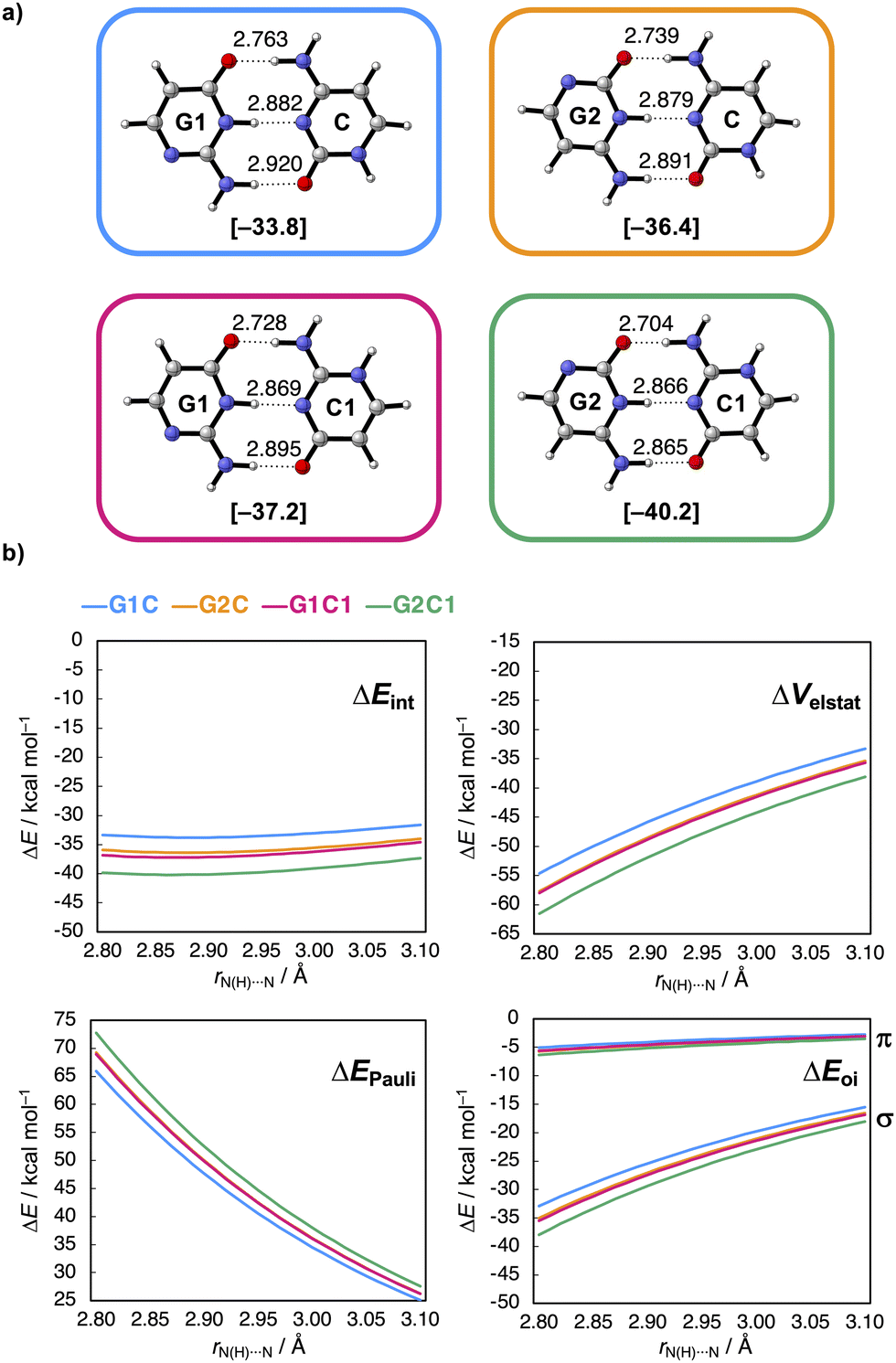
Uit de berekeningen van de onderzoekers bleek dat als je deze stikstof- en NH-groep twee plekjes in de ring opschuift, zodat ze respectievelijk dichter bij de C=O- en NH2-groep terechtkomen, de waterstofbruggen sterker worden (zie afbeelding): ze maten een vrij groot verschil in interactie-energie van 6,5 kcal/mol. Dat komt doordat de lading zich na het verschuiven ophoopt in de moleculen, wat op zijn beurt weer zorgt voor sterkere elektrostatische én σ-orbitaalinteracties tussen de moleculen.
Het geeft aanwijzingen voor twee observaties: allereerst lijken de elektrostatische interacties dus niet alleen maar afhankelijk van de frontier-atomen, maar ook van atomen die verder weg liggen. Ten tweede zijn de moleculaire orbitalen gedelokaliseerd over het complete molecuul, waardoor hun energieën dus worden beïnvloed wanneer je de ruggengraat van het molecuul aanpast. Met dit concept kun je het ontwerp van biomoleculen en waterstofbrugsystemen slimmer aanpakken.
Nieuwland, C. et al. (2024) PCCP 26, DOI: 10.1039/D3CP05244C










Nog geen opmerkingen